比较基因组学流程
cnblogs 2024-08-01 16:45:01 阅读 61
1、OrthoFinder 教程:用于比较基因组学的系统发育直系学推断
1.1 orthofinder介绍
OrthoFinder是一种快速、准确和全面的比较基因组学分析工具。它可以找到直系和正群,为所有的正群推断基因树,并为所分析的物种推断一个有根的物种树。OrthoFinder还为比较基因组分析提供全面的统计数据。OrthoFinder使用简单,只需运行一组FASTA格式的蛋白质序列文件(每个物种一个)。
1.2 Orthofinder安装
建议使用conda安装,简单方便,不用担心其他依赖和glibc版本的问题
1.创建一个工作目录。打开终端并运行以下命令:
<code>mkdir ~/orthofinder_tutorial
cd ~/orthofinder_tutorial
2.下载最新版本的 OrthoFinder(如果您使用 Bioconda,您仍需要执行此步骤来获取示例数据集)
wget https://github.com/davidemms/OrthoFinder/releases/latest/download/OrthoFinder.tar.gz
如果你没有安装 wget,你可以试试 curl:
curl -L -O https://github.com/davidemms/OrthoFinder/releases/latest/download/OrthoFinder.tar.gz
或者去GitHub发布页面下载OrthoFinder:https ://github.com/davidemms/OrthoFinder/releases
3.解压包,cd到OrthoFinder目录
tar xzvf OrthoFinder.tar.gz
cd OrthoFinder/
4.打印orthofinder帮助文件
orthofinder -h
5.在示例数据集上运行 OrthoFinder
orthofinder -f ExampleData/
你将会得到以下结果:
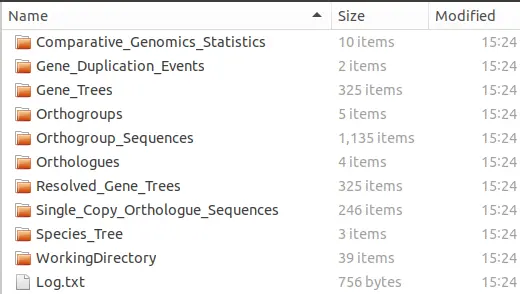
1.3 orhtofinder的使用示例
对一组模型生物进行系统基因组分析:小鼠、人类、青蛙、斑马鱼、日本河豚
在https://www.ensembl.org/,寻找相关基因[2]
OrthoFinder 需要输入你感兴趣的物种中所有蛋白质编码基因的氨基酸序列。每个物种的序列应该在一个单独的文件中,文件扩展名为“.fa”、“.faa”、“.fasta”、“.fas”或“.pep”。当一个物种的基因组被测序并可用时,会执行两个主要步骤,即组装和注释。组装是将单个读数拼接到基因组序列中。注释是对基因组组装中感兴趣的特征的识别,例如蛋白质编码基因。因此,我们需要的文件通常位于名为“注释”的部分中。在 Ensembl 的右侧,在“基因注释”下单击“下载 FASTA”。如图:

单击“pep”文件夹(其中包含肽序列),然后将文件“Homo_sapiens.GRCh38.pep.all.fa.gz”下载到创建的文件夹中。
重复这一步骤下载其他物种序列信息,之后使用<code>gunzip *.gz解压相关文件。
为加快运行时间,使用orthofinder提供的python脚本,提取每个基因的最长的转录变体
for f in *fa ; do python ~/orthofinder_tutorial/OrthoFinder/tools/primary_transcript.py $f ; done
你将会得到primary_transcripts文件夹,之后运行
nohup orthofinder -f primary_transcripts/ &
运行时间比较长,你将会得到以下结果

一些重要概念[1、3]
- Species-specific orthogroup: 一个仅来源于一个物种的直系同源组
- Single-copy orthogroup: 在直系同源组中,每个物种里面只有一个基因。我们会用单拷贝直系同源组里的基因推断物种树以及其他数据分析。
- Unassigned gene: 无法和其他基因进行聚类的基因。
- G50和O50,指的是当你直系同源组按照基因数从大到小进行排列,然后累加,当加入某个组后,累计基因数大于50%的总基因数,那么所需要的直系同源组的数目就是O50,该组的基因树就是G50.
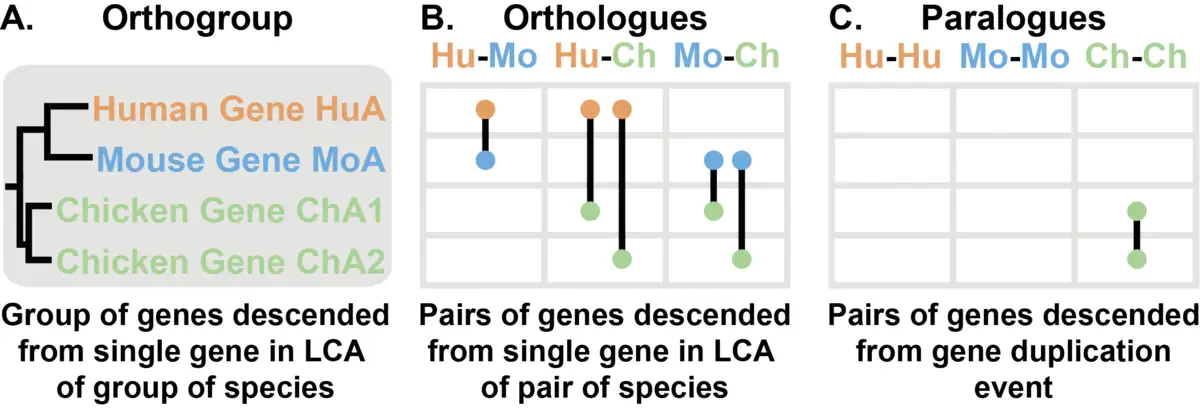
Orthogroups, Orthologs 和 Paralogs 这三个概念推荐看图理解。
1.4 结果文件解读[3]
运行标准OrthoFinder会产生一系列文件:直系同源组、直系同源、基因树、物种树、基因复制事件、比较基因组学数据。结果文件在所分析物种的.fa文件目录下。

(1)Orthogroups 直系同源组目录[2]
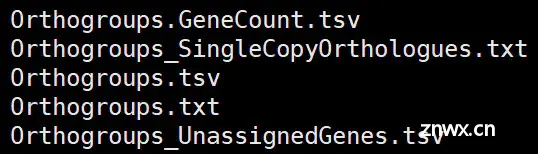
Orthogroups.tsv:用制表符分隔的文件,每一行包含属于单个直系同源组的基因。每个物种的直系同源组的基因单独排成一列。
Orthogroups.txt: 类似于Orthogroups.tsv,只不过是 OrthoMCL的输出格式。
Orthogroups_UnassignedGenes.tsv:用制表符分隔开的文件,格式上与Orthogroups.tsv相同,但包含的是未分配到任何直系同源组的基因。
Orthogroups_SingleCopyOrthologues.txt:每个物种正好包含一个基因的直系同源群列表,即它们包含一对一的直系同源物。 这种直系同源组非常适合进行种间比较和种树推断。
Orthogroups.GeneCount.tsv:格式同Orthogroups.tsv。每一行为一个直系同源组,每一列是每个物种每个直系同源组的基因数目。
(2)Orthologues 直系同源目录
在这个目录中,每个物种都有一个子目录。该子目录又包括每个物种对的比较文件。直系同源可以是一对一、一对多或多对多,这取决于基因复制事件。文件中的每一行都包含一个物种中的基因,而该基因是另一物种中该基因的直系同源物,并且每一行都被交叉引用到包含这些基因的直系同源组中。
(3)Comparative_Genomics_Statistics 比较基因组学数据目录[2]

Statistics_Overall.tsv:用制表符分隔开的文件,其中包含有直系同源组分析的常规统计信息。(总体统计信息)
Statistics_PerSpecies.tsv:用制表符分隔开的文件,内容与Statistics_Overall.tsv相似,但分物种。(分物种统计信息)
Duplications_per_Orthogroup.tsv:制表符分隔开的文件,该文件给出每个直系同源组中标识的复制项的数量。
Duplications_per_Species_Tree_Node.tsv:制表符分隔开的文件,该文件给出了沿物种树的每个分支发生的复制次数。
Orthogroups_SpeciesOverlaps.tsv:制表符分隔开的文件,包含每个物种对共享的直系同源群数目。
OrthologuesStats_?-to-?.tsv:制表符分隔开的文件,包含矩阵。这些矩阵给出了每对物种之间一对一,一对多和多对多关系的直系同源物数量。
(4)Gene_Duplication_Events 基因复制事件目录
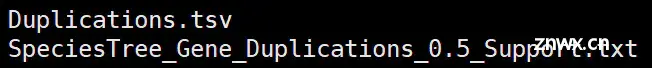
Duplications.tsv:制表符分隔的文件,其中列出了所有基因复制事件,这些事件是通过检查每个直系同源群基因树的每个节点来标识的。
SpeciesTree_Gene_Duplications_0.5_Support.txt:提供了物种树分支上上述复制的总和。 它是newick格式的文本文件。 每个节点/物种名称后面的数字是在导致节点/物种的分支上发生的具有至少50%支持的基因复制事件的数量。
(5)Gene_Trees 基因树目录

为每个直系同源群推断的系统发育树。
(6)Orthogroup_Sequences 直系同源组序列目录
每个直系同源群的FASTA文件,给出了每个直系同源群中每个基因的氨基酸序列。
(7)Resolved_Gene_Trees 解析的基因树目录
为每个直系同源组推断出有根的系统发育树,使用OrthoFinder复制损失合并模型进行解析。
(8)Single_Copy_Orthologue_Sequences 单拷贝直系同源组序列目录
与直系同源组序列目录相似的文件。每个物种一对一的直系同源组。
(9)Species_Tree 物种树目录

SpeciesTree_rooted.txt:从所有直系同源组推断出的STAG物种树,包含内部节点上的STAG支持值,并以STRIDE(Species Tree Root Inference from Duplication Events)为根。STAG(Species Tree from All Genes)是一种从所有基因推测物种树的算法,不同于使用单拷贝的直系同源基因进行进化树构建。
SpeciesTree_rooted_node_labels.csv:同上,但是节点具有标签,以允许其他结果文件交叉引用物种树中的分支/节点(例如基因复制事件的位置)。
本节参考文献:
- 「基因组学」使用OrthoFinder进行直系同源基因分析
- OrthoFinder-用于比较基因组学的系统发育直系学推断
- orthofinder结果解读
2.系统发育树的构建
利用orthofinder寻找单拷贝基因构建系统发育树
声明
本文内容仅代表作者观点,或转载于其他网站,本站不以此文作为商业用途
如有涉及侵权,请联系本站进行删除
转载本站原创文章,请注明来源及作者。