Nat Commun系列|如何像搭积木一样去搭建你自己的病理AI模型框架|专题总结·24-08-30
罗小罗同学 2024-09-11 17:01:32 阅读 61
小罗碎碎念
前情铺垫
今天的第一篇推文更偏向理论知识,分享了多篇综述,帮助大家快速了解病理切片染色标准化和虚拟染色的内容。
那么这期推文则是补充第一篇推文没有涉及的部分——染色标准化如何作为预处理流程出现在整体的框架中——准备了三篇Nature Communication,够有诚意了吧,哈哈。
其实搭建模型就像搭建积木一样,你完全可以把不同功能的模块视为一块块积木,那么我在最后给大家准备了数个病理染色标准化/虚拟染色的“积木”,然后试着模仿这三篇NC去做一个替换,搭建你自己的模型!!
一、基于回归的深度学习:病理切片图像中连续性分子生物标志物的精准预测

一作&通讯
| 角色 | 姓名 | 单位(中文翻译) |
|---|---|---|
| 第一作者 | Omar S. M. El Nahhas | 德累斯顿工业大学医学系,德国 |
| 通讯作者 | Jakob Nikolas Kather | 德累斯顿工业大学医学系,德国 |
文献概述
这篇文章报道了一种基于回归的深度学习方法,能够从病理切片图像中准确预测连续性的分子生物标志物,为计算病理学和精准医疗提供了新的工具。
研究团队开发并评估了一种自监督的、基于注意力机制的弱监督回归方法,直接从11,671张涵盖九种癌症类型的患者的图像中预测连续的生物标志物。研究发现,使用回归方法显著提高了生物标志物预测的准确性,并且与已知临床相关区域的对应性比分类方法更好。在结直肠癌患者的大型队列中,基于回归的预测分数比基于分类的分数具有更高的预后价值。该研究提供的开源回归方法为计算病理学中的连续生物标志物分析提供了一个有希望的替代方案。
文章还讨论了数字病理学的最新进展,包括直接从全切片图像(WSI)预测基因变化和基因表达模式的研究。研究指出,尽管大多数现有的深度学习方法限于分类问题,但许多生物标志物的真实值是连续的,通常在用于深度学习模型之前会被二值化。文章提出,回归分析可能是一种更适合的方法,因为它能够更好地处理这些连续值。
研究结果表明,回归模型在预测同源重组缺陷(HRD)评分方面的表现优于分类模型和Graziani等人提出的现有回归方法。此外,回归模型在预测与肿瘤微环境中的关键生物过程相关的生物标志物方面也显示出了优越的性能,包括肿瘤细胞增殖、基质分数和免疫细胞相关的生物标志物。
最后,文章强调了回归模型在预测肿瘤微环境中的生物标志物方面的优势,包括更好的对应已知临床相关区域的预测和在结直肠癌中改善生存预测的能力。研究还指出,尽管取得了进展,但仍有局限性,比如实验范围有限,且没有优化训练模型的超参数。未来的研究可能会扩展到更广泛的癌症类型和临床目标,同时考虑如何处理连续生物标志物的噪声和不确定性。
重点关注
Fig. 3展示了CAMIL(Contrastively-Clustered Attention-based Multiple Instance Learning)分类方法与CAMIL回归方法在预测肿瘤微环境中连续生物过程生物标志物方面的比较。
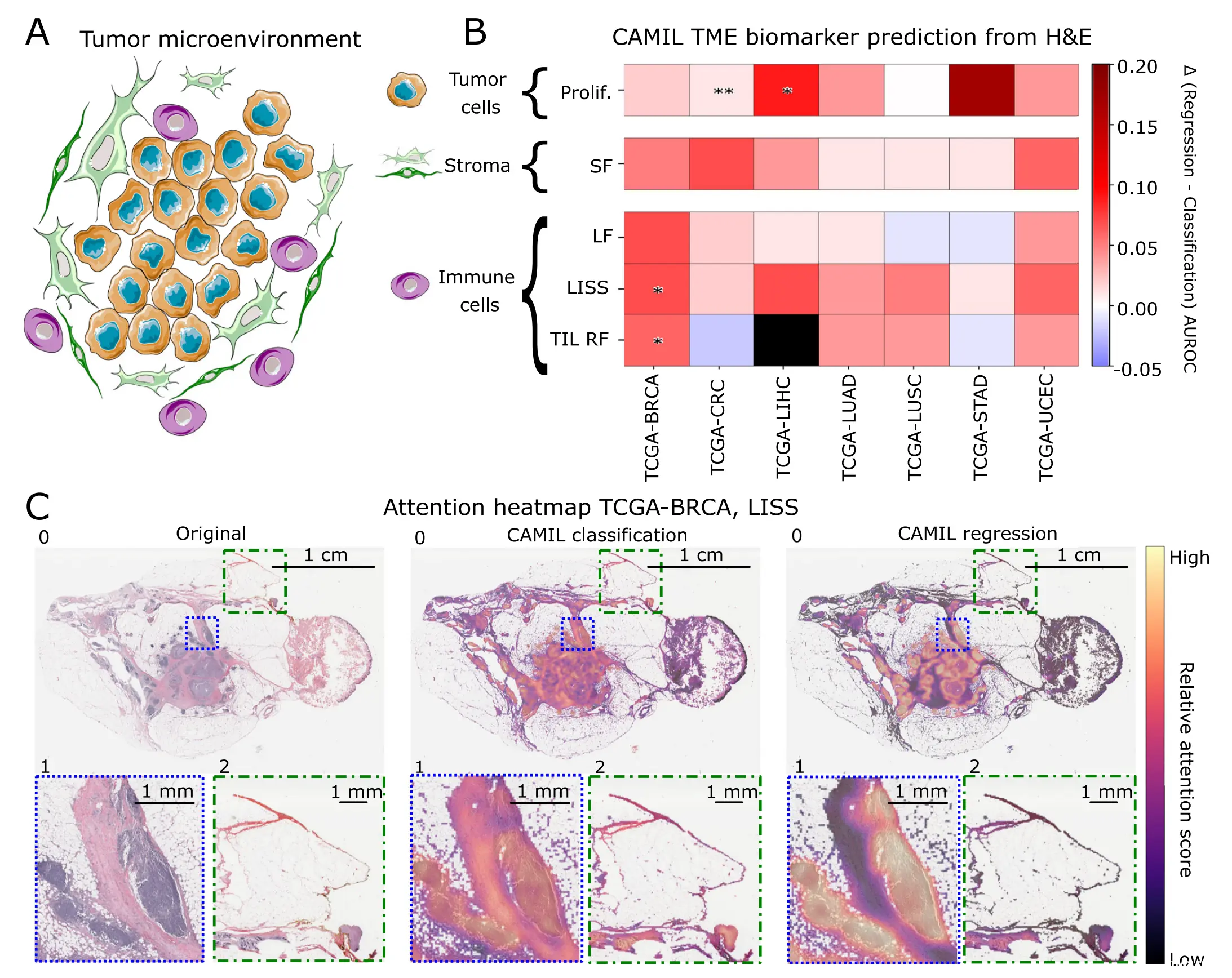
以下是对图3各部分的分析:
A部分:简化的肿瘤微环境(TME)图示,分析的主要焦点包括肿瘤细胞、基质和免疫细胞。
B部分:热图显示了CAMIL回归模型与CAMIL分类模型在五个生物过程生物标志物(肿瘤浸润性淋巴细胞区域比例TIL RF、增殖Prolif.、白细胞比例LF、淋巴细胞浸润签名得分LISS和基质分数SF)的接收者操作特征曲线下面积(AUROC)的差异。这些生物标志物在多种癌症类型上进行了测试,包括乳腺癌(BRCA)、结直肠癌(CRC)、肝细胞癌(LIHC)、肺腺癌(LUAD)、肺鳞状细胞癌(LUSC)、胰腺癌(PAAD)、胃癌(STAD)和子宫内膜癌(UCEC)。所有这些癌症类型的数据都来自癌症基因组图谱(TCGA)项目,并使用了地点意识分层。热图中较高的正值表明CAMIL回归模型的性能更优。星号表示成对的双尾DeLong测试统计显著性(α = 0.0167)。
C部分:代表性的注意力热图来自TCGA-BRCA测试集的一张幻灯片。图像0显示了整个幻灯片,突出了图像1中的一个诊断区域。图像2表示了一个可能包含非必要诊断信息的区域。对于原始幻灯片、使用CAMIL分类的注意力热图和使用CAMIL回归的注意力热图(针对LISS生物标志物),这种序列被重复。注意力得分较高的区域对模型的决策更为关键。源数据作为源数据文件提供。该图的部分内容使用了Servier Medical Art的图片绘制。
总体而言,Fig. 3强调了CAMIL回归模型在预测肿瘤微环境中关键生物标志物方面相较于CAMIL分类模型的优势,特别是在统计显著性和模型决策的相关区域对应性方面。
代码&数据
预处理管道代码链接:
链接:https://github.com/KatherLab/end2end-WSI-preprocessing/releases/tag/v1.0.0-preprocessing作用:这部分代码用于处理整张幻灯片图像(Whole Slide Images, WSIs),是图像分析流程中的第一步,为后续的特征提取和模型训练做准备。
分类管道代码链接:
链接:https://github.com/KatherLab/marugoto/releases/tag/v1.0.0-classification作用:该代码实现了CAMIL分类模型,用于根据病理图像预测生物标志物的分类标签,例如是否存在某种基因突变。
回归管道代码链接:
链接:https://github.com/KatherLab/marugoto/releases/tag/v1.0.0-regression作用:此代码实现了CAMIL回归模型,用于预测生物标志物的连续数值,比如基因表达水平或者肿瘤细胞的增殖分数。
分类和回归注意力热图代码链接:
链接:https://github.com/KatherLab/highres-WSI-heatmaps/releases/tag/v1.0.0-heatmaps作用:这部分代码用于生成和展示模型的注意力热图,帮助理解模型在做出预测时重点关注图像的哪些区域。
癌症基因组图谱(TCGA)数据集链接:
链接:https://portal.gdc.cancer.gov/作用:TCGA提供了大量的癌症基因组数据,包括基因组测序数据、病理图像等,这些数据在本研究中用于训练和验证深度学习模型。
临床蛋白质组肿瘤分析联盟(CPTAC)数据集链接:
链接:https://proteomics.cancer.gov/dataportal作用:CPTAC提供了蛋白质组学数据,这些数据可以与病理图像联合使用,以增强对癌症生物标志物的理解和预测。
癌症基因组图谱(TCGA)和CPTAC的分子数据链接:
链接:https://www.cbioportal.org/作用:cBioPortal是一个可视化和分析癌症基因组数据的平台,研究者可以利用这个平台整合和分析来自TCGA和CPTAC的分子数据。
DACHS研究的生物标志物数据请求链接:
链接:https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001113.v1.p1作用:DACHS研究提供了结直肠癌的生物标志物数据,这些数据对研究者来说是有价值的资源,但需要通过申请获得访问权限。
DACHS研究的联系信息链接:
链接:http://dachs.dkfz.org/dachs/kontakt.html作用:提供了DACHS研究的联系信息,研究者可以通过这个链接获取DACHS研究的幻灯片和生物标志物数据的访问。
这些代码和数据集是文章研究工作的基础,使得研究的可重复性和透明度得以保证,同时也为其他研究者在相同或相关领域的研究提供了便利。
二、深度学习算法在尿路上皮癌FGFR基因变异预测中的开发与应用

一作&通讯
| 角色 | 姓名 | 单位(中文) |
|---|---|---|
| 第一作者 | Albert Juan Ramon | 强生研发有限公司,数据科学与数字健康,美国加州圣地亚哥 |
| 通讯作者 | Kristopher A. Standish | 强生研发有限公司,数据科学与数字健康,美国加州圣地亚哥 |
文献概述
这篇文章报道了一种基于深度学习的组织病理学图像分析算法的开发和临床试验部署,该算法能够高效地预测尿路上皮癌患者FGFR基因变异,从而加速患者筛选并减少分子测试的需求。
研究背景:在肿瘤治疗中,准确识别肿瘤中的基因变异(如成纤维细胞生长因子受体,FGFR)对于使用靶向治疗至关重要。然而,分子测试可能因时间和组织需求量而延迟患者治疗。
AI算法开发:研究者开发了一种基于AI的生物标记检测算法,使用超过3000张H&E染色的全切片图像(WSIs),这些图像来自晚期尿路上皮癌患者。算法优化了高灵敏度以避免排除符合试验条件的患者。
算法验证:在350名患者的数据集上验证了算法,实现了0.75的曲线下面积(AUC),在88.7%的灵敏度下特异性为31.8%,并预计减少了28.7%的分子测试需求。
算法部署:研究者成功地将该系统部署在一个包含89个全球研究临床站点的非干预性研究中,并展示了其在优先/取消分子测试资源和在药物开发及临床环境中节省成本方面的潜力。
临床应用:FGFR靶向治疗虽然改善了临床护理,但由于成本高、周转时间长,分子测试作为标准护理的广泛应用仍然较慢。AI算法的部署有助于快速筛选患者,提高临床试验的效率。
技术细节:算法使用了公共数据存储库、商业来源和内部临床试验的数据集进行训练和验证,并在多个独立大型数据集上进行了稳健的验证。
结果:算法在独立数据集上的表现一致,且在真实世界的临床试验数据上也显示出了良好的泛化能力。
讨论:研究展示了AI在临床环境中的潜力,尤其是在提高患者护理效率、减少筛查负担和提供快速的临床洞察方面。
结论:这项工作为精准医疗提供了一个步骤,通过快速、可行的临床洞察,增加了患者获得有效、靶向治疗的机会。
文章强调了AI在医疗领域的应用潜力,特别是在提高临床试验患者筛选效率和减少医疗成本方面的重要性。
重点关注
Fig. 3展示了可解释性分析的结果,其中包括了对正确预测(true positive)和错误预测(true negative)的尿路上皮癌(MIBC)组织切片图像(WSIs)的分析:
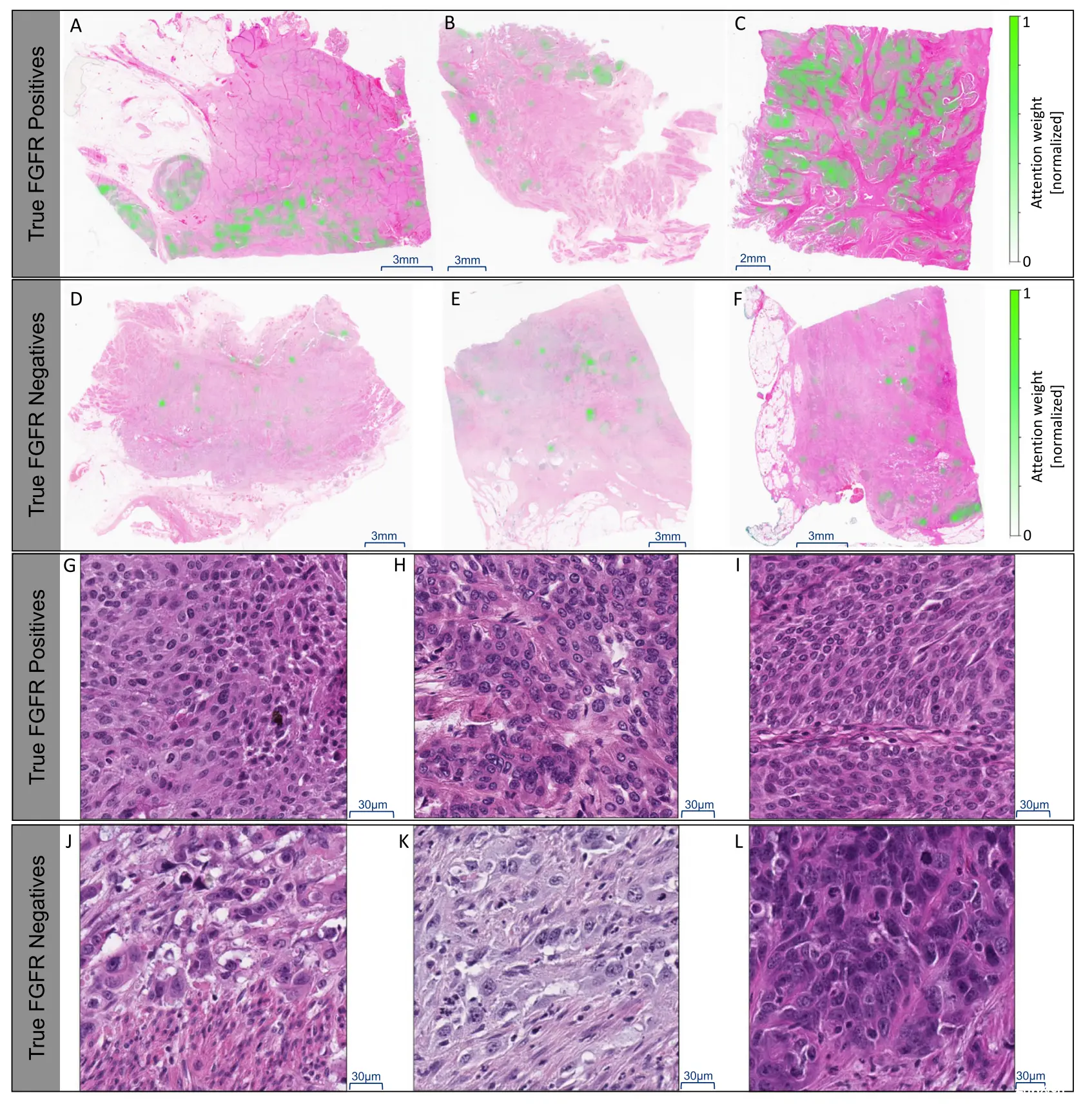
A至C:展示了来自BLC3001研究的随机选择的正确预测的WSIs,这些图像上有表示模型注意力权重的热图(绿色)。这些权重被标准化为1,每个面板右侧的颜色条映射到标准化权重,所有图像共用相同的颜色条。颜色越亮的绿色区域表示模型认为对做出预测更重要的区域(更高的注意力权重)。注意,无论幻灯片被预测为FGFR阳性或阴性,算法都会在用于推理的所有瓦片上产生注意力权重。
D至F:展示了错误预测的WSIs的热图示例。这些图像上的热图显示了模型认为不重要的区域,即这些区域的注意力权重较低。
G至I:在40倍放大倍数下,从A至C展示的正确预测中得分最高的瓦片图像,显示出更多实体肿瘤细胞,细胞分级较低至中等,这与之前的观察结果33一致。
J至L:从D至F展示的错误预测中得分最高的瓦片图像,显示出相对更多分散的高细胞学分级的肿瘤细胞。
简而言之,Fig. 3中的图像和热图提供了模型在进行FGFR状态预测时所关注图像区域的可视化,这些区域可能与肿瘤细胞的形态学特征有关。正确预测的图像倾向于集中在低至中等分级的实体肿瘤细胞区域,而错误预测的图像则更多地集中在高细胞学分级的分散肿瘤细胞区域。这种分析有助于理解模型的预测行为,并可能为病理学家提供有关FGFR阳性肿瘤特征的额外见解。
代码&数据
代码链接:
pyTorch ResNet34 Network
作用:文章中使用的卷积神经网络(CNN)的初始权重来源于这个预训练的ResNet34网络,用于图像特征提取。
CODAIT Deep-Histopath
作用:文章中提到,用于训练和推理的多实例学习(multi-instance learning)管道部分基于这个开源项目。这个项目提供了处理和分析全切片图像(WSIs)的基础代码。
数据集:
The Cancer Genome Atlas (TCGA) Consortium
作用:文章中使用了来自TCGA的数据集,这些数据被用于算法的开发阶段,特别是在训练和验证深度学习模型时。
Janssen R&D Clinical Studies
包括以下研究:
NCT03955913NCT03390504NCT03473743 作用:这些临床研究提供了用于算法训练、验证和部署的H&E染色的全切片图像和相关的分子测试结果。
其他资源:
StainTools
作用:用于处理和标准化H&E染色图像的染色效果,用于数据预处理阶段。
补充信息:
Janssen and Johnson FGFR Device Review作用:这个链接提供了用于数据分析和图形生成的预测和源代码,供读者进一步了解研究的方法和结果。
这些代码链接和数据集为研究提供了必要的工具和资源,使得研究人员能够开发、训练和验证他们的深度学习模型,并最终在临床试验中部署使用。
三、人工智能在区分子宫内膜癌亚型中的应用

一作&通讯
| 角色 | 姓名 | 单位 |
|---|---|---|
| 第一作者 | Amirali Darbandsari | 不列颠哥伦比亚大学电子与计算机工程系 |
| 第一作者(共同) | Hossein Farahani | 不列颠哥伦比亚大学生物医学工程系 |
| 第一作者(共同) | Maryam Asadi | 不列颠哥伦比亚大学生物医学工程系 |
| 通讯作者 | Ali Bashashati | 不列颠哥伦比亚大学生物医学工程系 |
文献概述
这篇文章利用人工智能技术对子宫内膜癌(Endometrial Cancer, EC)的组织病理学图像进行分析,发现了一种新的具有较差预后的亚型,这可能有助于改善患者的治疗决策。
研究背景:子宫内膜癌有四种分子亚型,它们具有强烈的预后价值和治疗意义。最常见的亚型是NSMP(无特定分子特征),在排除其他三种分子亚型的特定特征后被指定,包括具有异质性临床结果的患者。
研究目的:本研究使用基于AI的组织病理学图像分析来区分p53abn和NSMP EC亚型,并识别出具有明显较差无进展生存期和疾病特异性生存率的NSMP EC患者亚群(称为“p53abn-like NSMP”)。
研究方法:研究者利用深度学习模型来分析来自不同中心的子宫内膜癌患者的组织病理学图像。他们训练了一个深度卷积神经网络(CNN)来识别肿瘤和非肿瘤区域,然后使用多实例学习(MIL)模型来区分p53abn和NSMP EC的图像。
研究结果:在发现队列中的368名患者以及两个独立验证队列中的290和614名患者中,研究发现p53abn-like NSMP组的患者与NSMP组相比,具有更高的基因组不稳定性,表现为更多的拷贝数异常。此外,研究还发现p53abn-like NSMP与NSMP相比,在临床、病理和分子特征上存在差异。
研究意义:这项工作展示了AI在检测预后不同且传统分子或病理标准无法识别的EC亚群方面的能力,从而改进了基于图像的肿瘤分类。研究结果仅适用于女性。
研究结论:AI方法可以用于识别那些在常规组织病理学评估中无法区分的具有侵袭性疾病进程的NSMP EC患者亚群,这可能有助于更精确地进行风险分层和治疗决策。
这篇文章强调了AI在医学图像分析中的潜力,特别是在提高癌症患者预后评估和治疗个性化方面的应用前景。
重点关注
Fig. 1 展示了基于人工智能的组织病理学图像分析的工作流程,具体步骤如下:
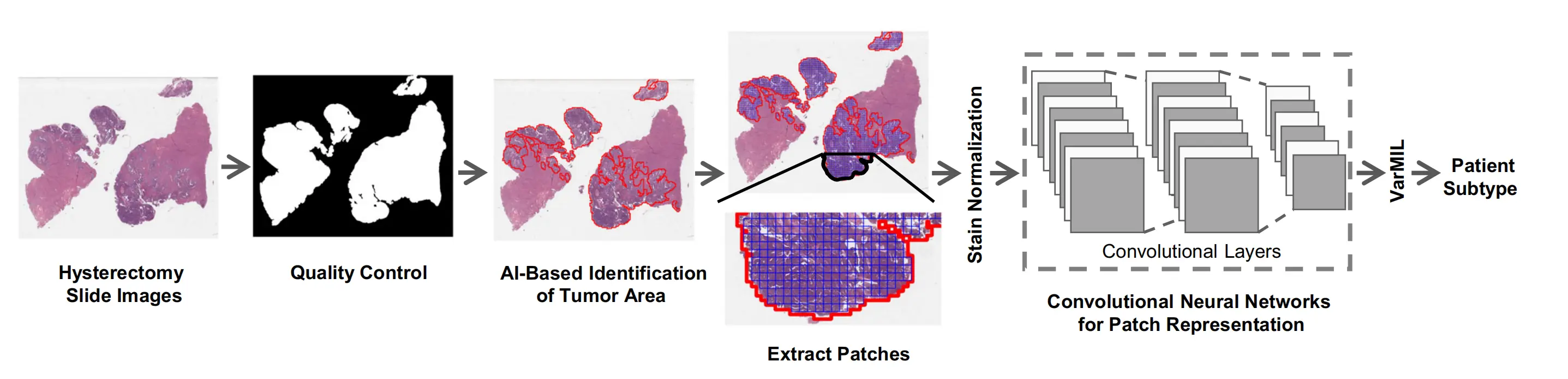
质量控制:首先使用质量控制框架HistoQC81生成一个掩膜,该掩膜仅包含组织区域,并去除伪影。肿瘤区域识别:接着,训练一个AI模型来识别组织病理学幻灯片中的肿瘤区域。图像分割与标准化:然后,将图像分割成小的块(patches),并进行标准化处理以消除颜色变化。深度学习特征提取:将标准化后的图像块输入到深度学习模型中,以获得块级别的表示(特征)。多实例学习预测:最后,使用基于多实例学习(VarMIL)的模型来预测患者的亚型。
整个流程是一个从原始的组织病理学图像到最终亚型预测的自动化过程,涉及图像预处理、特征学习和分类预测等关键步骤。
代码&数据
代码链接:
https://github.com/AIMLab-UBC/EC-p53abnlike-AIclassifier
作用:这是文章中使用的AI模型的代码仓库,它包含了训练和应用深度学习模型以区分子宫内膜癌不同亚型所需的代码。这个链接提供了实现研究结果的算法和模型的访问,允许其他研究人员或开发人员复现、学习和进一步开发该技术。
数据集:
The Cancer Genome Atlas (TCGA) project 数据集
作用:TCGA项目提供了用于研究的基因组数据,包括子宫内膜癌的基因组和外显子测序数据。文章中使用这些数据来定义和验证子宫内膜癌的分子亚型。 British Columbia (BC) cohort 数据集
作用:这是来自不列颠哥伦比亚省的独立验证队列,用于验证AI模型的预测准确性和稳定性。 Cross Canada (CC) cohort 数据集
作用:这是来自加拿大26个医院的多中心数据集,用于进一步验证AI模型的泛化能力和在不同中心数据上的适用性。 Tübingen University Women’s Hospital 数据集
作用:来自德国图宾根大学医院的数据,用于发现队列的构建,为AI模型的训练和初步验证提供数据。
四、其余染色标准化相关的算法推荐
https://github.com/HaoyuCui/WSI_Normalizer
https://github.com/xindubawukong/Vahadane
https://github.com/MEDAL-IITB/Fast_WSI_Color_Norm
https://github.com/ChenYuhang243/stain-normalizer
https://github.com/KatherLab/end2end-WSI-preprocessing/tree/v1.0.0-preprocessing
https://github.com/pegahs1993/Stain-to-Stain-Translation
https://github.com/Peter554/StainTools
下一篇: AI公文写作哪家强?2024年4款最强AI公文写作工具推荐-附深度测评
本文标签
声明
本文内容仅代表作者观点,或转载于其他网站,本站不以此文作为商业用途
如有涉及侵权,请联系本站进行删除
转载本站原创文章,请注明来源及作者。